
スケールを横断したさまざまな特性を満たすプロセスの設計や新しいエネルギー貯蔵・変換材料の開発などにおいて、実験科学の手法に加えて、計算化学やデータ科学、自律実験系などを融合させた次世代の材料開発手法の構築が求められています。しかしながら、第一原理計算などを用いた物質・材料シミュレーションは実効的な時空間スケールの計算を実行するには膨大な時間を要するという課題がありました。私たちはさまざまな材料の原子間相互作用を機械学習力場で表現し、計算機が判別した未知の構造に対してのみ、第一原理計算によって新たに取得した訓練データを用いて相互作用モデルを再構築する自律的な学習アルゴリズムを考案しました。その結果、従来と比較して計算速度を2~4桁ほど向上させることが可能となりました。これを触媒活性や電解液のイオン伝導率などの計算に適用し、実験との連携をはかることで材料設計を加速させていきます。
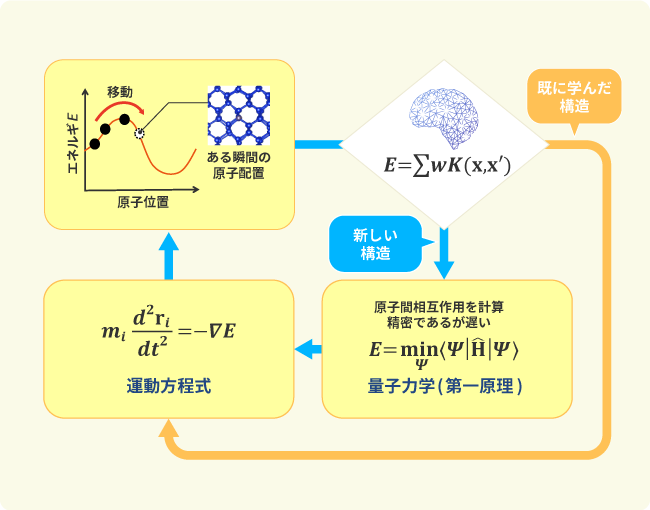
シミュレーション中に自ら学ぶデータ駆動化学計算技術