
Developing new energy storage and conversion materials as well as processes assembling the materials ranged over a wide length scale into a functional device requires advanced designing methods that combine computational physics, data science, and autonomous experimental systems together with empirical science methods. Conventional simulation methods, such as first-principles (FP) calculations, however, have severe limitations because of trade-off relationships between required computational time, accuracy and robustness. To solve the problem, we have devised an autonomous learning algorithm that generates interatomic interaction models of various materials automatically during FP simulations on the fly. The algorithm enables the machine to judge whether a structure appearing in the simulation is out of database or not. If the structure is judged to be new, the machine automatically gets FP data and reconstructs the interaction model. Otherwise, the structure is updated by using the interaction model. In this manner, most of FP calculations are bypassed, and two to four orders of magnitude faster simulations are realized. We will apply this new approach to calculations of ionic conductivity of electrolytes and activity of catalysts. Collaborations of the advanced simulations and experiments will pave the way to highly efficient materials design.
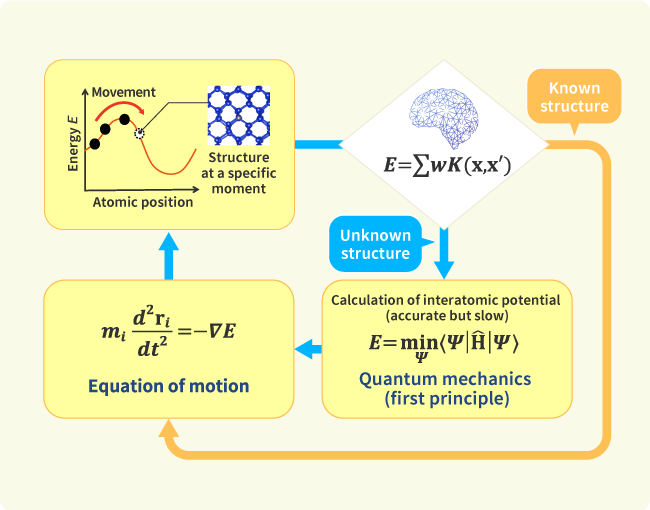
Data-driven chemical calculation technology that learns on its own during simulations