
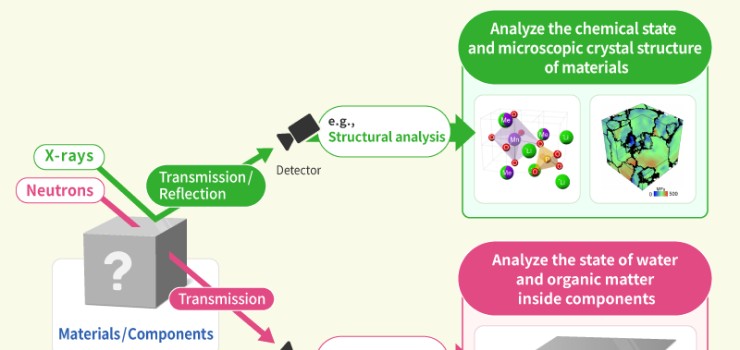
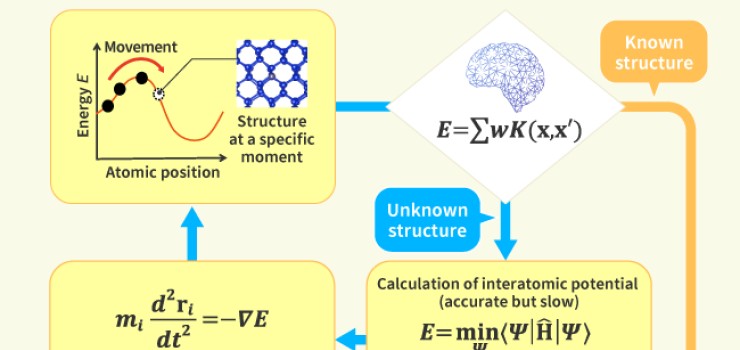
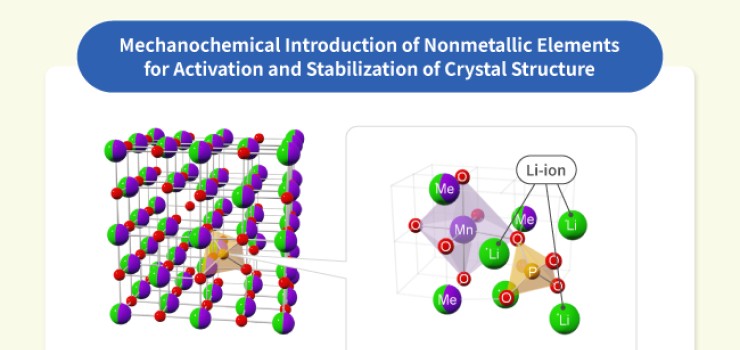
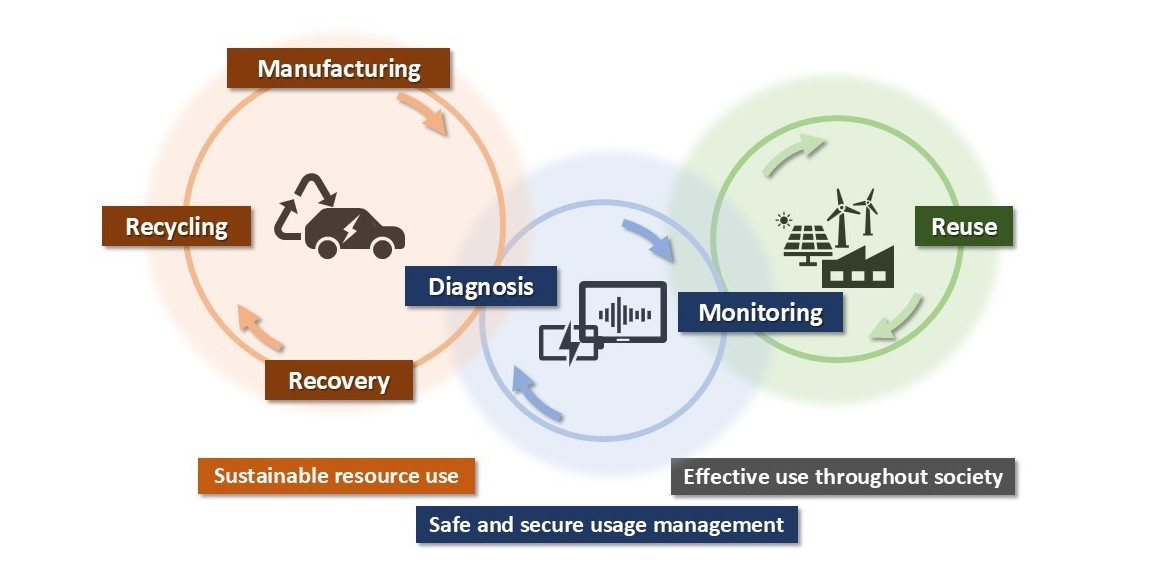
We engage in research and development into the materials that will create breakthroughs in response to increasingly complex social demands, such as the environmental performance and economic rationality required by next-generation automobiles and energy systems. As part of our efforts to create energy storage and conversion materials, in addition to nano-interface reaction design and reaction control technologies, we also advance research and development into technologies that analyze and elucidate phenomena related to material structures and reactive processes at multiple scales using X-rays, light, and quantum beams. Moreover, we work with microfabrication technologies, manufacturing technologies including additive fabrication that integrates material processes and molding, material design at the atomic and molecular level based on computational science, and process development that enables the synthesis of these materials. In addition, we are actively engaged in materials informatics, autonomous and automated experiments, and other forms of fusion research with computer science as a means of dramatically improving the efficiency of materials development.
Nanomaterials, Mathematical Physics and Matter Physics, Metal Materials Properties, Inorganic and Coordination Chemistry, Energy Chemistry, Chemical Reaction and Process System Engineering, Catalyst and Resource Chemical Process, Analytical Chemistry, Quantum Beam Science, Environmental Materials and Recycle Technology






-
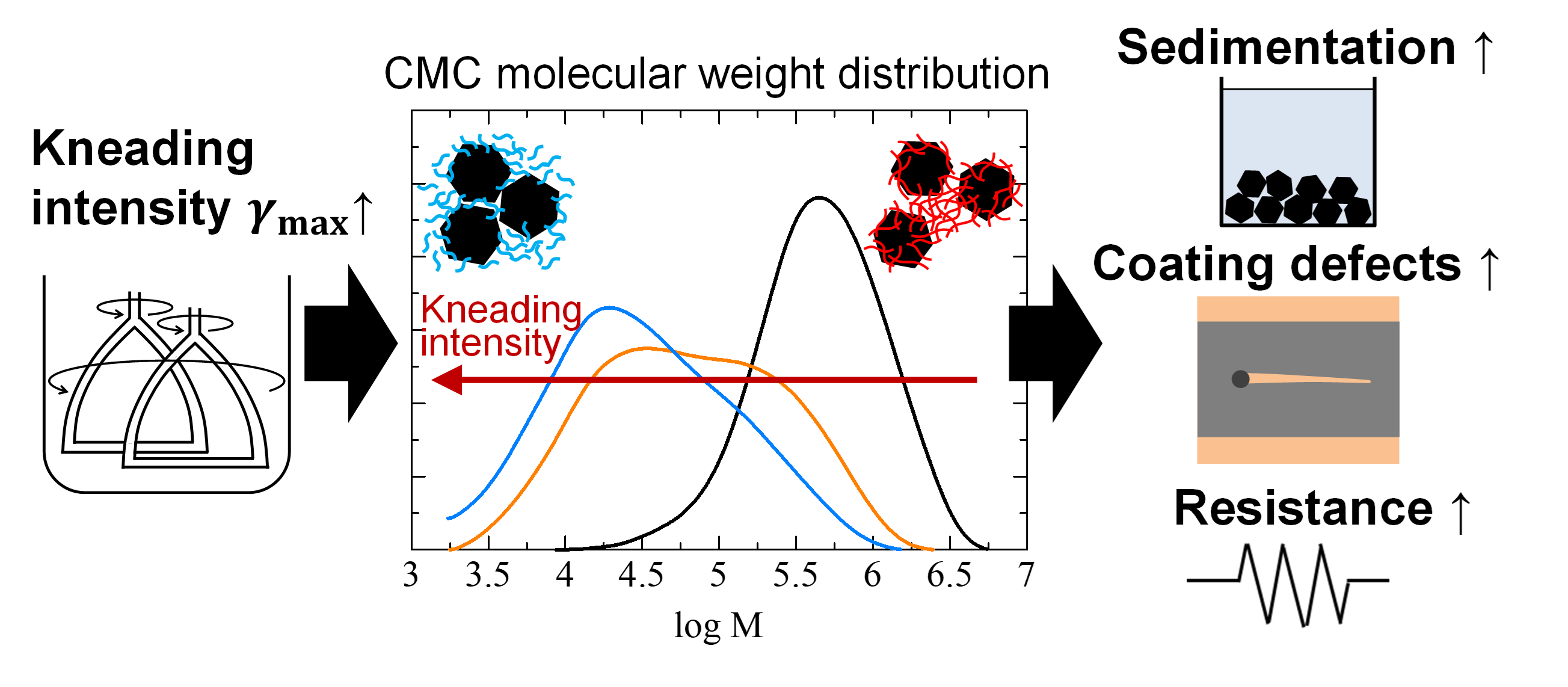
Jul 22, 2026 High-Solid-Content Kneading Degrades Carboxymethyl Cellulose as a Dispersant and Destabilises Graphite Anode Slurries
- Makino, S., Ishii, M., Kawauchi, S., Sasaki, M., Akimoto, Y., Nakamura, H.
- Journal of Colloid and Interface Science
- Vol. 708, (2025), 139769.
-
Jul 13, 2026 Electrostatic Dry Coating Strategies for Through-Plane Control of Electronic Pathways in Lithium-Ion Electrodes
- Yonaga, A., Matsunaga, T.
- Small Methods
- Vol. 10, No. 9, (2026), e02213.
-
Jul 8, 2026 Photocatalytic CO2 reduction with a semiconductor/metal complex hybrid system: Toward visible light and water utilization
- Morikawa, T., Suzuki, T. M., Yamaguchi, Y., Kudo, A.
- Journal of Photochemistry & Photobiology C: Photochemistry Reviews
- Vol. 66, (2026), 100735.
-
Jun 18, 2026 Water Generation in the Mesoporous Catalyst Layers of Automotive Fuel Cells: Insights from Operando Small-Angle Neutron Scattering
- Harada, M., Hasegawa, N., Kato, A., Higuchi, Y., Kato, S., Song, F., Iwase, H., Takata, S.
- Chemical Engineering Journal
- Vol. 525, (2025), 170753.
-
Jun 9, 2026 Fluorene-Based Capacity Recovery Reagent for LiFePO₄ Batteries
- Shibata, Y., Mikita, R., Kondo, H.
- ACS Sustainable Resource Management
- Vol. 3, (2026), 96–104.